


Muskelspinneatrofi manifesterer seg tidligbarndom. De første symptomene kan oppstå så tidlig som 2-4 måneders alder. Dette er en arvelig sykdom som er preget av at nerveceller gradvis dør i hjernestammen.

Spinal muskelatrofi kan utvikle seg ved:
- den første typen: akutt (Verdig-Hoffmann-form);
- den andre typen: mellomliggende (infantil, kronisk);
- til den tredje typen: Kugelberg-Velander-form (kronisk, ung).
Tre typer av samme sykdom oppstår,på grunn av forskjellige mutasjoner av samme gen, ifølge eksperter. Muskulær spinalatrofi er en autoimmun sykdom som oppstår når to recessive gener blir arvet, en fra hver av foreldrene. Mutasjonsstedet er lokalisert på kromosom 5. Den er til stede i hver 40 person. Genet er ansvarlig for å kode et protein som sikrer eksistensen av motoriske nevroner i ryggmargen. Hvis denne prosessen blir forstyrret, dør nevronene.
Du kan mistenke et problem i løpet avsvangerskap. Hvis et barn utvikler spinal muskelatrofi av type 1, noteres ofte en treg og sen fosterbevegelse under graviditet. Etter fødselen kan legene diagnostisere generalisert muskelhypotensjon.

Men dette er ikke alle tegnene som kan værebestemt spinal muskelatrofi. Symptomer som er karakteristiske for type 1 av denne sykdommen inkluderer svakhet i interkostale muskler. I dette tilfellet ser brystet til babyene flate ut.
I de første månedene av livet lider disse barna ofte av luftveisinfeksjoner, lungebetennelse og hyppig aspirasjon.
Du kan bestemme sykdommen hvis du undersøkeret barn. Det er en rekke kliniske tegn som spesialister kan bestemme at babyen har arvelig spinal muskelatrofi. Diagnostikk inkluderer:
- biokjemisk analyse av blod (det vil vise en svak økning i aldolase og kreatinfosfokinase);
- elektromyografisk undersøkelse (rytmen i utspenningen vil vitne om skade på de fremre hornene i ryggmargen);
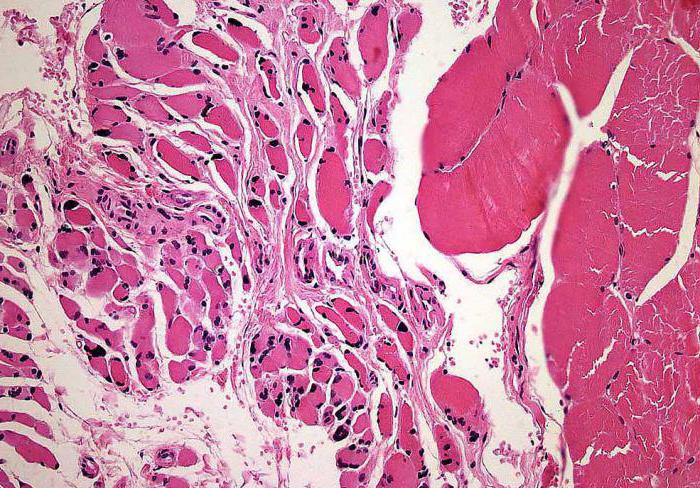
- histologisk undersøkelse av skjelettmuskulatur (klynger av avrundede småfibre oppdages).
Mikroskopi av ryggmargen (dets fremre horn)viser at det er degenerative forandringer i motorkjernene i kraniale nerver. Sfærisk hevelse og / eller rynking av motorceller, mikroglial eller astrocytisk spredning, kromatolyse. Disse fenomenene er ledsaget av utseendet på tette glialfibre.
Det er viktig å utføre differensialdiagnostikk,for å utelukke organisk aciduria, medfødte eller strukturelle myopatier, for eksempel pemalin, myotubular myopati eller sentralstammesykdom.

Proksimale muskler er de første som liderunderekstremiteter. Nederlagsprosessen er på stigende linje. Med progresjonen av sykdommen blir musklene i strupehodet og svelget påvirket. Det hender at sykdommen dukker opp litt senere. Men grensen er 6 måneder. Barn begynner til og med å løfte hodet og rulle over, men de setter seg aldri.
De fleste babyer dør av hjertesykdom,respirasjonssvikt eller infeksjoner det første leveåret. Mer enn 70% av barna dør før 2 år. Cirka 5% av disse barna overlever til 5 års alder.
Tynne fingre indikerer sykdommenfremtredende fascikulær skjelving i musklene i de proksimale lemmer, ved fingertuppene. Disse babyene har en periode med normal utvikling. De i riktig alder begynner å holde hodet, setter seg. Men det er ikke snakk om uavhengig gåing.
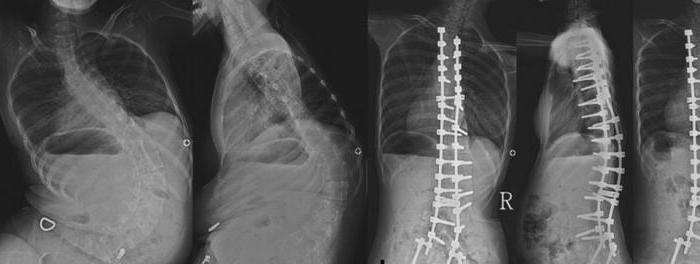
Barn kan suge og svelge normalt, tidligSpedbarns respirasjonsfunksjon er ikke nedsatt. Men over tid begynner muskelsvakhet å utvikle seg. I en eldre alder vises svelgplager, en nesetone i stemmen oppstår. En av de vanligste komplikasjonene hos pasienter med en betydelig forventet levealder er skoliose.
Til tross for at barnet i de første månedene kanutvikler seg normalt, forverres situasjonen over tid. I en alder av to år forsvinner senrefleksene i lemmene hos barn. Parallelt svekkes de interkostale musklene. Som et resultat blir brystet flatet ut, og motorisk utvikling er forsinket.
Bare 25% av barna har fått diagnosenspinal muskelatrofi, kan sitte eller stå på egen hånd med støtte. Senere stadier av sykdommen er preget av utseendet på kyfoskopiose. Mange barn dør mellom 1 og 4 år. De vanligste dødsårsakene er lungebetennelse eller muskelskader i luftveiene.
Når du etablerer en diagnose av spinal muskelatrofi, er det nødvendig å utføre differensialdiagnose med medfødte eller strukturelle myopatier, atonisk-astatisk type cerebral parese.
Mer enn 70% av pasientene lever i mer enn to år. Gjennomsnittlig levealder for slike barn er omtrent 10-12 år.
Pasienter har vanskeligheter med å gå, løpe,å komme seg opp fra en hukestilling og behovet for å klatre opp trapper. Det er verdt å huske at de kliniske manifestasjonene av denne sykdommen i denne formen ligner den progressive Beckers dystrofi.
Proksimale armer og skulderbelteblir berørt bare noen få år etter de første manifestasjonene av sykdommen. Over tid er brystet deformert, fascikulær skjelving i hendene og ukontrollert sammentrekning av forskjellige muskelgrupper vises. Samtidig avtar senreflekser og deformiteter i bein begynner å utvikle seg. Brystet, føttene, ankelleddene endres, spinal skoliose vises.
Kugelberg-Welander sykdom erspinal muskelatrofi hos voksne. Sykdommen gjennom tidene har noe komplisert pasienters liv, men fører ikke til deres alvorlige funksjonshemming. Det går ganske sakte. Derfor lever de fleste med denne diagnosen i voksen alder.
Før du starter behandlingen, er det viktig ådifferensialdiagnose med forskjellige muskeldystrofier, glykogenose type 5 og strukturelle myopatier. Det er en metode der spinalatrofi kan bestemmes. Dette er en direkte DNA-diagnose. Det er nødvendig fordi de kliniske manifestasjonene av Kugelberg-Welander sykdom kan variere.
I omtrent 50% av tilfellene taper pasientermuligheten til å gå fra 12 år. Samtidig utvikler muskelsvakhet bare med alderen. Ledd hypermobilitet og kontrakturer observeres, og det er en økt risiko for brudd.
Svakhet og muskelsammentrekninger i spedbarnetalder, håndskjelv og forsinket motorisk utvikling, bør varsle både leger og foreldre. Slike barn stiller ikke opp. Bare en fjerdedel av alle pasienter kan stå med støtte. Barna er innesperret i rullestol.
Men det vanskeligste er å definere sykdommenKugelberg-Welander. I en fjerdedel av pasientene er muskelhypertrofi faktisk uttalt. Derfor kan de feilaktig bli diagnostisert med muskeldystrofi i stedet for spinal muskelatrofi. Årsakene til denne sykdommen ble etablert i 1995, da det var mulig å identifisere det muterende SMN-genet. Nesten alle pasienter har en homozygot sletting av SMN7, preget av tap av to telomere kopier av dette genet og intakte sentromere kopier.

I noen tilfeller kan leger bare lindre tilstanden til pasienter ved hjelp av fysioterapi. Spesielle ortopediske enheter bidrar til å forbedre livskvaliteten.
Spesiell oppmerksomhet bør rettes mot ernæring av slikesyk. Legemidler som kan forbedre metabolismen er ofte foreskrevet. Men dette er ikke alt som kan gjøres hvis muskelatrofi i ryggraden er bekreftet. Behandlingen består i utnevnelse av gymnastikk. Fysisk aktivitet for slike pasienter er viktig. De er nødvendige for å forbedre muskelfunksjonen og bygge masse. Det er sant at belastningen skal beregnes av legen. I dette tilfellet vil kroppsøving ha en gunstig effekt på hele kroppen som helhet. Den generelle styrkende effekten vil være åpenbar. Foreskrevet kan være terapeutiske eller hygieniske morgenøvelser, øvelser i vannet, eller bare en generell styrkende massasje.


