


Kas spinal atrofisi erken dönemde kendini gösterirçocukluk. İlk belirtiler 2-4 ay kadar erken ortaya çıkabilir. Bu, sinir hücrelerinin yavaş yavaş beyin sapında ölmesiyle karakterize kalıtsal bir hastalıktır.

Spinal kas atrofisi şu şekilde gelişebilir:
- ilk tip: akut (Verdig-Hoffmann formu);
- ikinci tip: ara (çocuksu, kronik);
- üçüncü tip: Kugelberg-Velander formu (kronik, genç).
Aynı hastalığın üç türü ortaya çıkar,uzmanlara göre, aynı genin farklı mutasyonları nedeniyle. Müsküler spinal atrofi, her bir ebeveynden bir tane olmak üzere iki resesif gen kalıtsal olarak meydana gelen otoimmün bir hastalıktır. Mutasyon yeri 5 kromozomu üzerinde bulunur. Her 40 kişide var. Gen, omurilikteki motor nöronların varlığını sağlayan bir proteinin kodlanmasından sorumludur. Bu işlem bozulursa, nöronlar ölür.
Sırasında bir sorundan şüphelenebilirsingebelik. Bir çocuk tip 1 spinal müsküler atrofi geliştirirse, hamilelik sırasında halsiz ve geç bir fetal hareket sıklıkla görülür. Doğumdan sonra, doktorlar genelleştirilmiş kas hipotansiyonunu teşhis edebilir.

Ancak bunlar olabilecek tüm işaretler değilbelirlenmiş spinal müsküler atrofi. Bu hastalığın tip 1'in karakteristik belirtileri interkostal kasların zayıflığını içerir. Bu durumda, bebeklerin göğsü düzleşmiş görünüyor.
Yaşamın ilk aylarında, bu çocuklar sıklıkla solunum yolu enfeksiyonları, zatürree ve sık sık aspirasyon geçirir.
Eğer muayene ederseniz, hastalığı belirleyebilirsiniz.bir çocuk Bebeğin kalıtsal spinal müsküler atrofiye sahip olduğunu belirleyen uzmanların belirleyebileceği çok sayıda klinik bulgu vardır. Teşhis şunları içerir:
- kanın biyokimyasal analizi (aldolaz ve kreatin fosfokinazda hafif bir artış gösterecektir);
- elektromiyografik çalışma (bordürün ritmi, omuriliğin ön boynuzlarına verilen zarar hakkında tanıklık edecektir);
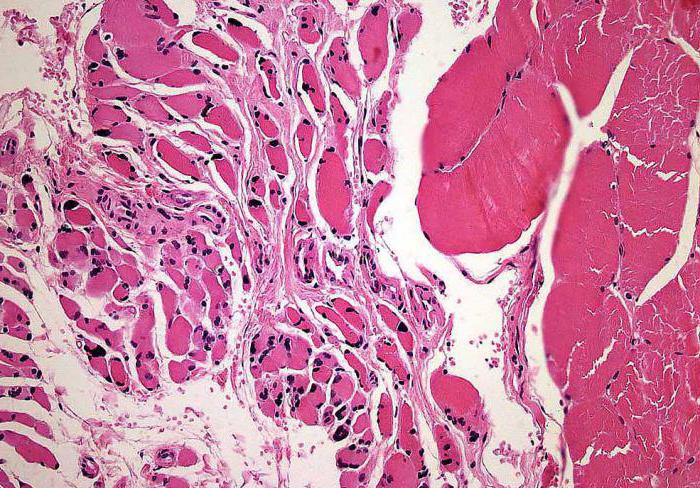
- iskelet kaslarının histolojik muayenesi (yuvarlak küçük liflerin kümeleri tespit edilir).
Omuriliğin mikroskobu (ön boynuzları)Kranial sinirlerin motor çekirdeğinde dejeneratif değişiklikler olduğunu göstermektedir. Motor hücrelerinin küresel şişmesi ve / veya kırışması, mikroglial veya astrositik proliferasyon, kromatoliz gözlenir. Bu fenomenlere yoğun glial liflerin ortaya çıkması eşlik eder.
Ayırıcı tanılama yapmak önemlidir.organik asidüri, konjenital veya yapısal miyopatileri, örneğin pemalini, miyotubüler miyopatiyi veya merkezi kök hastalığını dışlamak için.

Proksimal kaslar ilk acı çeken kaslardır.alt uzuvlar. Yenilgi süreci artan bir çizgidedir. Hastalığın ilerlemesi ile, gırtlak ve farenks kasları etkilenir. Hastalığın biraz sonra ortaya çıkması ile olur. Ancak sınır 6 aydır. Çocuklar başlarını kaldırmaya ve yuvarlanmaya bile başlayabilir, ancak asla oturmazlar.
Çoğu bebek kalp hastalığından ölür,solunum yetmezliği veya yaşamın ilk yılında önceki enfeksiyonlar. Çocukların% 70'inden fazlası 2 yaşından önce ölmektedir. Bu çocukların yaklaşık% 5'i 5 yaşına kadar hayatta kalmaktadır.
İnce parmaklar hastalığı gösterirproksimal uzuvların kaslarında parmak uçlarında ortaya çıkan fasiküler titreme. Bu bebeklerin normal gelişim dönemleri vardır. Doğru yaşta başlarını tutmaya, oturmaya başlarlar. Ancak bağımsız yürüyüşten söz edilmiyor.
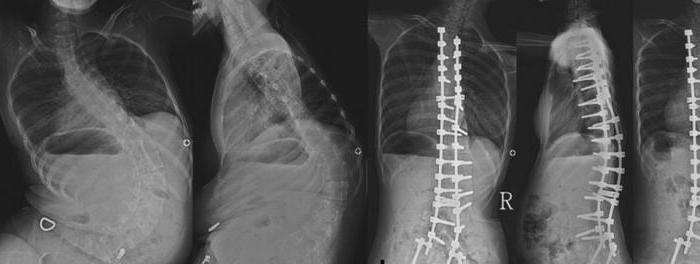
Çocuklar normalde emebilir ve yutabilirler.Bebeğin solunum fonksiyonu bozulmaz. Ancak zamanla kas güçsüzlüğü ilerlemeye başlar. Daha büyük yaşlarda, yutma bozuklukları ortaya çıkar, sesin burun tonu oluşur. Yaşam beklentisi anlamlı olan hastalarda en sık görülen komplikasyonlardan biri skolyozdur.
İlk aylarda çocuğunnormal olarak gelişir, zamanla durum kötüleşir. İki yaşına geldiğinde, çocuklarda ekstremitelerin tendon refleksleri kaybolur. Paralel olarak, interkostal kaslar zayıflar. Sonuç olarak, göğüs düzleştirilir, motor gelişiminde bir gecikme vardır.
Çocukların sadece% 25'i teşhis edildispinal müsküler atrofi, kendileri oturabilir veya destekle ayakta durabilirler. Hastalığın geç aşamaları, kifoskolyozun ortaya çıkması ile karakterizedir. Birçok çocuk 1 ile 4 yaşları arasında ölmektedir. En yaygın ölüm nedenleri pnömoni veya solunum kaslarına verilen zarardır.
Spinal müsküler atrofiyi teşhis ederken, serebral palsi atonik-astatik bir formu olan konjenital veya yapısal miyopatiler ile ayırıcı tanı koymak gerekir.
Hastaların% 70'inden fazlası iki yıldan fazla yaşıyor. Bu çocukların ortalama yaşam beklentisi yaklaşık 10-12 yıldır.
Hastalar yürüme, koşma,çömelme pozisyonundan kalkmak ve merdiven çıkma ihtiyacı. Bu hastalığın bu formdaki klinik belirtilerinin, Becker'in ilerleyen distrofisine benzer olduğunu hatırlamakta fayda var.
Proksimal Kol ve Omuzhastalığın ilk belirtilerinden sadece birkaç yıl sonra etkilendi. Zamanla, göğüs deforme olur, ellerin fasiküler titremesi ve çeşitli kas gruplarının kontrolsüz kasılması ortaya çıkar. Bu durumda, tendon refleksleri azalır ve kemik deformiteleri ilerlemeye başlar. Göğüs, ayaklar, ayak bileği eklemleri değişiyor, spinal skolyoz ortaya çıkıyor.
Kugelberg-Velander hastalığıyetişkinlerin spinal kas atrofisi. Yıllar boyunca, hastalık hastaların yaşamlarını biraz karmaşıklaştırır, ancak ciddi sakatlığa yol açmaz. Oldukça yavaş ilerliyor. Bu nedenle, bu teşhisi olan çoğu insan yetişkinliğe yaşar.
Tedaviye başlamadan önce,çeşitli kas distrofileri, tip 5 glikojenoz ve yapısal miyopatiler ile ayırıcı tanı. Spinal atrofiyi belirleyebileceğiniz bir yöntem vardır. Bu doğrudan bir DNA teşhisidir. Bu gereklidir, çünkü Kugelberg-Velander hastalığının klinik belirtileri değişebilir.
Vakaların yaklaşık% 50'sinde hastalar kaybeder12 yaşından itibaren yürüme yeteneği. Aynı zamanda, kas güçsüzlüğü sadece yaşla birlikte ilerler. Hipermobilite ve eklemlerin kasılması gözlenir, artmış kırık riski ortaya çıkar.
Bebekte zayıflık ve kas kasılmalarıyaş, el titremesi ve motor geriliği hem doktorları hem de ebeveynleri uyarmalıdır. Bu çocuklar ayağa kalkmaz. Tüm hastaların sadece dörtte biri destekle ayakta durabilir. Çocuklar tekerlekli sandalyeyle sınırlıdır.
Ama belirlenmesi en zor şey hastalıkKugelberg-Welander. Gerçekten de, hastaların dörtte birinde kas hipertrofisi belirgindir. Bu nedenle, yanlışlıkla spinal müsküler atrofi ile değil, kas distrofisi ile teşhis edilebilir. Bu hastalığın nedenleri, mutasyona uğrayan SMN genini tanımlamanın mümkün olduğu 1995 yılında kuruldu. Hemen hemen tüm hastalar, bu genin iki telomerik kopyasının ve bozulmamış centromere kopyalarının kaybı ile karakterize edilen homojen bir SMN7 delesyonu yaşarlar.

Bazı durumlarda, doktorlar fizyoterapi yardımıyla hastaların durumunu hafifçe hafifletebilir. Yaşam kalitesini artırmak özel ortopedik cihazlara izin verir.
Bu türlerin beslenmesine özellikle dikkat edilmelidir.Hastalar. Genellikle metabolizmayı artırabilecek reçeteli ilaçlar. Ancak spinal müsküler atrofi doğrulanırsa yapılabileceklerin hepsi bu değildir. Tedavi jimnastik atanmasından oluşur. Bu tür hastalar için fiziksel aktivite önemlidir. Kasların işleyişini iyileştirmek ve kütlelerini arttırmak için gereklidirler. Doğru, yük doktor tarafından hesaplanmalıdır. Bu durumda, beden eğitimi bir bütün olarak tüm organizma üzerinde faydalı bir etkiye sahip olacaktır. Onarıcı etkisi açıktır. Atanan terapötik veya hijyenik sabah egzersizleri, su egzersizleri veya sadece onarıcı bir masaj olabilir.


