


Spinalna mišićna atrofija se manifestuje u ranim fazamadetinjstvo. Prvi simptomi se mogu javiti već od 2-4 meseca života. Ovo je nasledna bolest koju karakteriše postepeno odumiranje nervnih ćelija u moždanom stablu.

Spinalna mišićna atrofija se može razviti:
- prvi tip: akutni (forma Verdig-Hoffmann);
- drugi tip: srednji (infantilni, hronični);
- treći tip: oblik Kugelberg-Welander (hronični, juvenilni).
Javljaju se tri vrste iste bolesti,prema mišljenju stručnjaka, zbog različitih mutacija istog gena. Spinalna mišićna atrofija je autoimuna bolest koja se javlja kada se nasleđuju dva recesivna gena, po jedan od svakog roditelja. Mesto mutacije se nalazi na hromozomu 5. Prisutan je kod svakih 40 ljudi. Gen je odgovoran za kodiranje proteina koji čini postojanje motornih neurona u kičmenoj moždini. Ako je ovaj proces poremećen, neuroni umiru.
Možete posumnjati na problem čak i tokomtrudnoća. Ako dete razvije spinalnu mišićnu atrofiju tipa 1, često se primećuje sporo i kasno kretanje fetusa tokom trudnoće. Nakon rođenja, lekari mogu dijagnostikovati generalizovanu hipotoniju mišića.

Ali ovo nisu svi znaci po kojima može postojatiutvrđena je spinalna mišićna atrofija. Simptomi karakteristični za tip 1 ove bolesti uključuju slabost interkostalnih mišića. Istovremeno, grudni koš beba izgleda spljošten.
U prvim mesecima života ova deca često boluju od respiratornih infekcija, upale pluća i česte aspiracije.
Možete odrediti bolest ako pregledatedete. Postoji niz kliničkih znakova po kojima stručnjaci mogu utvrditi da beba ima naslednu spinalnu mišićnu atrofiju. Dijagnostika uključuje:
- biohemijski test krvi (pokazaće blagi porast aldolaze i kreatin fosfokinaze);
- elektromiografska studija (ritam stokade će ukazati na leziju prednjih rogova kičmene moždine);
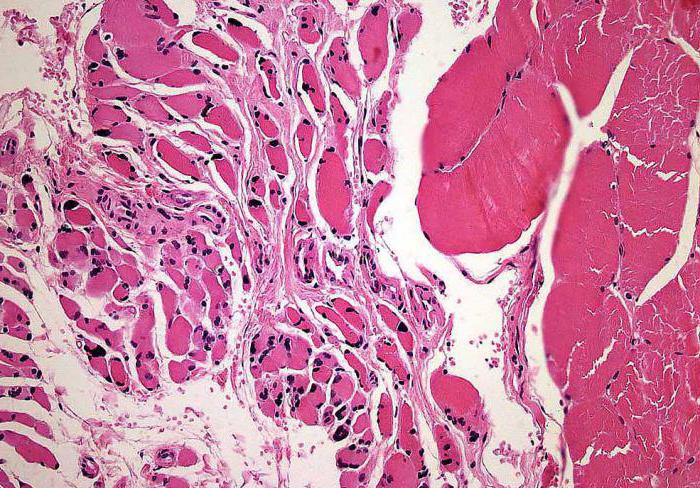
- histološki pregled skeletnih mišića (otkrivaju se klasteri zaobljenih malih vlakana).
Mikroskopija kičmene moždine (njeni prednji rogovi)pokazuje da postoje degenerativne promene u motornim jezgrima kranijalnih nerava. Uočeno je sferno oticanje i / ili skupljanje motornih ćelija, mikroglijalna ili astrocitna proliferacija, hromatoliza. Ove pojave su praćene pojavom gustih glijalnih vlakana.
Važno je sprovesti diferencijalnu dijagnostiku,da se isključe organske acidurije, urođene ili strukturne miopatije, na primer, pemalinska miopatija, miotubularna miopatija ili bolest centralnog štapića.

Proksimalni mišići su prvi koji pate.Доњи екстремитети. Proces poraza ide uzlaznom linijom. Kako bolest napreduje, zahvaćeni su mišići larinksa i ždrela. Dešava se da se bolest manifestuje malo kasnije. Ali starosna granica je 6 meseci. Deca mogu čak početi da podižu glavu i prevrću se, ali nikada ne sede.
Većina beba umire od srcarespiratorna insuficijencija ili prethodne infekcije u prvoj godini života. Više od 70% dece umire pre 2 godine. Oko 10% ove dece preživi do 5 godina.
Stanji prsti svedoče o bolesti,pojavni fascikularni tremor u mišićima proksimalnih ekstremiteta, u vrhovima prstiju. Ove bebe imaju period normalnog razvoja. U odgovarajućem dobu počinju da se drže za glavu, sede. Ali samostalno hodanje ne dolazi u obzir.
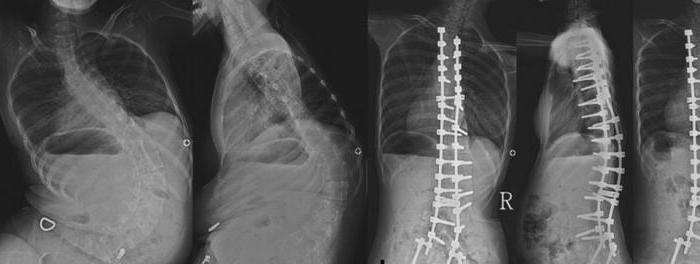
Deca mogu da sišu i gutaju normalno, ranou detinjstvu, respiratorna funkcija nije oštećena. Ali vremenom, slabost mišića počinje da napreduje. U starijoj dobi pojavljuju se poremećaji gutanja, pojavljuje se nazalni ton glasa. Jedna od najčešćih komplikacija kod pacijenata sa dugim životnim vekom je skolioza.
Uprkos činjenici da u prvim mesecima dete možerazvijaju normalno, situacija se vremenom pogoršava. Do dve godine, tetivni refleksi udova nestaju kod dece. Paralelno, interkostalni mišići slabe. Kao rezultat, grudni koš je spljošten i pojavljuje se kašnjenje u motoričkom razvoju.
Samo 25% dece kojoj je dijagnostikovanaspinalna mišićna atrofija, mogu samostalno sedeti ili stajati uz podršku. Kasnije stadijume bolesti karakteriše pojava kifoskolioze. Mnoga deca umiru između 1 i 4 godine. Najčešći uzroci smrti su pneumonija ili oštećenje respiratornih mišića.
Prilikom dijagnoze spinalne mišićne atrofije potrebno je sprovesti diferencijalnu dijagnostiku sa urođenim ili strukturnim miopatijama, atonično-astatskim tipom cerebralne paralize.
Više od 70% pacijenata živi duže od dve godine. Prosečan životni vek takve dece je oko 10-12 godina.
Pacijenti otežano hodaju, trče,ustajanje iz čučećeg položaja i potreba za penjanjem uz stepenice. Vrijedno je zapamtiti da su kliničke manifestacije ove bolesti u ovom obliku slične progresivnoj Beckerovoj distrofiji.
Proksimalne ruke i rameni pojassu pogođeni samo nekoliko godina nakon prvih manifestacija bolesti. Vremenom se grudni koš deformiše, pojavljuje se fascikularni tremor ruku i nekontrolisana kontrakcija različitih mišićnih grupa. Istovremeno, refleksi tetiva se smanjuju i deformiteti kostiju počinju da napreduju. Pojavljuju se promene u grudnom košu, stopalima, skočnim zglobovima, skolioza kičme.
Kugelberg-Welanderova bolest jespinalna mišićna atrofija kod odraslih. Dugi niz godina, bolest donekle komplikuje život pacijenata, ali ne dovodi do njihove teške invalidnosti. Napreduje prilično sporo. Zbog toga većina ljudi sa ovom dijagnozom živi do odraslog doba.
Pre početka lečenja važno je dadiferencijalna dijagnoza sa različitim mišićnim distrofijama, glikogenozom tipa 5 i strukturnim miopatijama. Postoji metod koji se može koristiti za određivanje atrofije kičme. Ovo je direktna DNK dijagnoza. Neophodno je jer kliničke manifestacije Kugelberg-Velanderove bolesti mogu varirati.
U oko 50% slučajeva pacijenti gubesposobnost hodanja od 12 godina. Istovremeno, slabost mišića samo napreduje sa godinama. Uočavaju se hipermobilnost i kontrakture zglobova, javlja se povećan rizik od preloma.
Slabost i kontrakcije mišića u detinjstvugodine, tremor ruku i odložen motorički razvoj, trebalo bi da upozori i lekare i roditelje. Takva deca ne stoje na nogama. Samo četvrtina svih pacijenata može da stoji sa podrškom. Deca su vezana za invalidska kolica.
Ali najteže je identifikovati bolestKugelberg-Welander. Zaista, kod četvrtine pacijenata izražena je hipertrofija mišića. Stoga im se može pogrešno dijagnostikovati mišićna distrofija, a ne spinalna mišićna atrofija. Uzroci ove bolesti ustanovljeni su 1995. godine, kada je bilo moguće identifikovati mutirajući SMN gen. Skoro svi pacijenti imaju homozigotnu deleciju SMN7, koju karakteriše gubitak dve telomerne kopije ovog gena i netaknute centromerne kopije.

U nekim slučajevima, lekari uz pomoć fizioterapije mogu samo malo da olakšaju stanje pacijenata. Njihov kvalitet života može se poboljšati posebnim ortopedskim aparatima.
Posebnu pažnju treba obratiti na ishranu takvihbolestan. Često se propisuju lekovi koji mogu poboljšati metabolizam. Ali to nije sve što se može učiniti ako je potvrđena spinalna mišićna atrofija. Lečenje se sastoji u imenovanju gimnastike. Fizička aktivnost za takve pacijente je važna. Potrebni su za poboljšanje funkcije mišića i izgradnju mišićne mase. Istina, opterećenje treba da izračuna lekar. U ovom slučaju, fizičko vaspitanje će imati blagotvorno dejstvo na celo telo u celini. Njegov opšti efekat jačanja će biti očigledan. Mogu se propisati terapeutske ili higijenske jutarnje vežbe, vežbe u vodi ili samo opšta masaža za jačanje.


