


El síndrome de Pfayffer es extremadamente rarouna enfermedad genética que ocurre en promedio en uno de cada 100,000 recién nacidos. Igualmente a menudo afecta a niños y niñas. El principal síntoma de la enfermedad es la coalescencia temprana de los huesos del cráneo en el proceso de embriogénesis, debido a lo cual el cerebro ya no puede desarrollarse normalmente.
La enfermedad fue descubierta por el famoso alemángenetista Rudolf Pfeiffer. En 1964, describió los síntomas de una enfermedad previamente desconocida, cuyos principales signos eran anormalidades en la estructura del cráneo y fusión de los dedos en los pies o las manos (sindactilia). El síndrome se observó en ocho pacientes de la misma familia, lo que indica la naturaleza genética de la enfermedad. Otros estudios han demostrado que el síndrome de Pfeiffer se transmite por un tipo autosómico dominante.
La ecografía puede detectar esta enfermedad en el feto ya en el segundo trimestre del embarazo. En la ecografía, se observan anomalías en el desarrollo del cráneo, los órganos internos y las extremidades.
En 1993, el genetista estadounidense Michael CohenPropuso clasificar el síndrome de Pfeiffer en tres tipos según la gravedad de los síntomas. Ahora su clasificación es generalmente aceptada y ampliamente utilizada en la literatura médica.
Los síntomas del síndrome de Pfayffer tipo 1 incluyenCraneosinostosis - una condición en la que una o más suturas craneales se fusionan prematuramente, formando un hueso holístico. Como resultado, se observan cambios en la forma del cráneo y características faciales anormales: hipertelorismo, orejas de implantación baja. Puede haber un deterioro de la visión, aumento de la presión intracraneal, paladar hendido. En el 50% de los pacientes, se observa pérdida de audición. La mayoría de las personas con este tipo de enfermedad tienen un nivel normal de inteligencia y no tienen anormalidades neurológicas.
У детей с типом 1 синдрома Пфайффера внешние signos, como dedos anchos y cortos en los brazos y las piernas, se observan casi siempre. La sindactilia es un síntoma posible, pero no necesario, para este tipo de patología.

Este tipo de síndrome se hereda de manera autosómica dominante.
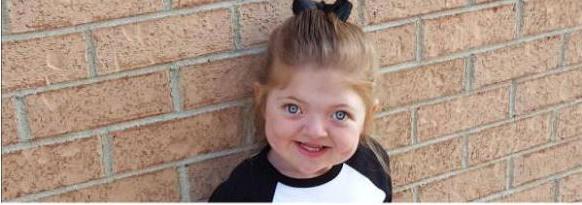
A continuación se muestra una foto del síndrome de Pfeiffer. La esperanza de vida en Rusia para las personas que padecen esta enfermedad excede los 60 años.
Para la enfermedad de tipo 2, el cráneo se caracteriza por unaforma de "trébol", la fusión de las vértebras, la sindactilia y el desplazamiento de los globos oculares hacia adelante (proptosis). Desde los primeros días de vida, aparecen graves trastornos neurológicos.
El tercer tipo de enfermedad se caracteriza aún máscraneosinostosis temprana El cráneo es anormalmente alargado y no forma un "trébol". Hay anomalías de los dientes, hipertelorismo, retraso mental, malformaciones de los órganos internos.
Lifetime en 2do y 3er tipola enfermedad varía de varios días a varios meses: los defectos de los órganos internos y los trastornos neurológicos no son compatibles con la vida. Las mutaciones responsables de estos dos tipos de síndrome de Pfeiffer ocurren esporádicamente y no son genéticamente heredadas.
El síndrome de Pfayffer se asocia con mutaciones del receptor 1factor de crecimiento de fibroblastos (FGFR1) en el cromosoma 8 o de crecimiento de fibroblastos receptor del factor de 2 (FGFR2) en el cromosoma 10. Los genes que afectan a la mutación responsable para el crecimiento normal de los fibroblastos, que desempeña un papel clave en el desarrollo de los huesos del cuerpo.

La medicina moderna aún no permite eliminarmutaciones en el genoma humano, incluso si se sabe en qué genes ha habido perestroika. Por lo tanto, para el síndrome de Pfeiffer, como para la mayoría de las otras enfermedades genéticas, el tratamiento es sintomático. Para los pacientes con tipo 2 y tipo 3, el pronóstico es desfavorable: numerosas anomalías del desarrollo no pueden corregirse ni por motivos médicos ni quirúrgicos.
El síndrome de Pfayffer tipo 1 es compatible con la vida.El síntoma principal de la enfermedad, la fusión prematura de los huesos del cráneo, se puede corregir quirúrgicamente. La operación se recomienda para niños hasta la edad de tres meses.

Además, la cirugía es efectiva en el caso de sindactilia simple. Es mejor comenzar el tratamiento a la edad de 18 a 24 meses, en el período de crecimiento activo de los dedos de manos y pies.


